


 搜索
搜索



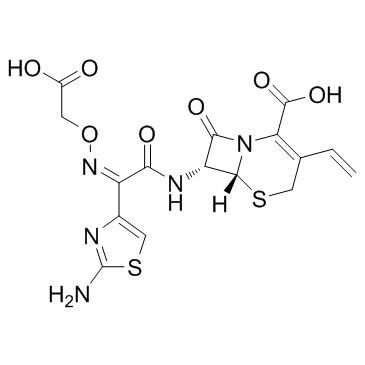
头孢克肟
-
中文名头孢克肟 三水合物
-
英文名Cefixime
-
中文别名头孢克肟
-
英文别名
Cefspan Oroken (6R,7R)-7-({(2-Amino-1,3-thiazol-4-yl)[(carboxymethoxy)imino]acetyl}amino)-8-oxo-3-vinyl-5-thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid 5-Thia-1-azabicyclo[4.2.0]oct-2-ene-2-carboxylic acid, 7-[[2-(2-amino-4-thiazolyl)-2-[(carboxymethoxy)imino]acetyl]amino]-3-ethenyl-8-oxo-, (6R,7R)- Saprax Suprax Cefixime CFIX MFCD03788802 fk027 Cefixim Unixime -
常用名头孢克肟
-
C A S号79350-37-1
-
密度1.9±0.1 g/cm3
-
沸点N/A
-
熔点218-225°C
-
化学式C16H15N5O7S2
-
结构式
1、 摩尔折射率:130.08
2、 摩尔体积(cm3/mol):302.7
3、 等张比容(90.2K):898.9
4、 表面张力(dyne/cm):77.7
5、 极化率(10-24cm3):51.57
-
闪点N/A
-
分子量453.450
-
精确质量453.041290
-
P S A238.05000
-
外观形状白色至灰白色粉末或灰白色晶体粉末
-
储存条件Hygroscopic, Refrigerator, Under Inert Atmosphere
-
稳定性
-
水溶解性
-
形态
-
H L B值
-
粘度
-
PH值
方法1:以7-ACA为原料。
7-ACA(18.7g,67mmol)悬浮于120ml水和120ml甲醇的混合溶液中,在-20℃下滴加氢氧化钠(5.5g,142mmo1)溶18ml水的溶液,在-10~-20℃下搅拌25min,用浓盐酸中和至Ph:7.5。在15℃下加入水杨醛(10.7g,87.6mmo1),然后搅拌1h。加入二苯基重氮甲烷(15.7g,80.9mmo1)在85ml乙酸乙酯的溶液,继续搅拌1h。在反应过程中,用1mol/L盐酸保持反应液的Ph值为4.0~4.5。反应液用550ml乙酸乙酯提取,提取液用盐水洗,减压浓缩。剩余物用石油醚浸渍,得29.6g化合物(I),收率78.1%,熔点97~98.5℃(分解)。化合物(I)(1.01g,2mmo1)溶于10ml二氯甲烷,在-30℃下加入五氯化磷(0.46g,2.2mmo1)和吡啶(0.176g,2.2mmo1),在-20~-30℃下搅拌1h。将反应液倾入冰水,分出有机层,用盐水洗,干燥,减压浓缩。得0.9g化合物(Ⅱ),收率85.7%,熔点180.5~182.0℃。
化合物(Ⅱ)(4.0g,7.6mmo1)溶于10ml二甲基甲酰胺,在冷却和搅拌下加入三苯膦(2.2g,8.4mmo1)。加毕,在室温下搅拌2h。将反应液倾入250ml异丙醇中,过滤收集沉淀(6.7g)。将该沉淀溶于10ml二氯甲烷和5ml水,加入17.4ml 36%甲醛水溶液。用10%碳酸钠水溶液调至Ph=9.0。在室温下搅拌1h后,用20ml二氯甲烷提取。提取液用盐水洗,干燥,减压浓缩。得3.0g化合物(Ⅲ),收率78.9%,熔点180.5~182℃(分解)。
化合物(Ⅲ)(18.96g,38.2mmo1)溶于135ml乙酸乙酯和34ml乙醇的混合液中,在室温下加入5.7ml浓盐酸,搅拌1.5h。过滤收集沉淀,用乙酸乙酯洗,干燥。得11。32g化合物(Ⅳ),收率69%,熔点172~173℃(分解)。
在-5~0℃和搅拌下,往二甲基甲酰胺(3.66g,50.1mmo1)和80ml四氢呋喃中,滴加三氯氧磷(7.7g,50.1mmo1),搅拌30min得到Vismeier试剂。在冰浴冷却下,加入化合物(V)(13.8g,41.8mmo1),再搅拌1h得到化合物(V)的活性溶液。在-20℃下,将该溶液加到化合物(Ⅳ)(15g,34.9mmo1)和N-(三甲基硅基)乙酰胺(MSA)(32g,244mo1)在150ml乙酸乙酯的溶液中,继续搅拌30min。加入乙酸乙酯和水,分出有机层,用5%碳酸氢钠溶液和盐水洗,干燥。减压浓缩,剩余物用异丙醇浸渍,得23.1g化合物(Ⅵ),收率97.5%
化合物(Ⅵ)(19.0g,28.0mmo1)溶于380ml甲醇,在室温下加入浓盐酸(11.6g,112mmo1),搅拌1h。用5%碳酸氢钠水溶液中和,减压浓缩。剩余物溶于乙酸乙酯后,用盐水洗,干燥。减压浓缩,剩余物用异丙醇浸渍,得15.3g化合物(Ⅶ),收率84.1%。
化合物(Ⅶ)(15.0g,23mmo1)溶于15ml苯甲醚,在冰浴冷却下,加入60ml三氟乙酸。加毕,在室温下搅拌80min。在搅拌下,将该反应液滴加到600ml异丙醇中。过滤收集产生的沉淀,溶于5%碳酸氢钠水溶液,用乙酸乙酯洗。用5%盐酸将该水溶液调至Ph=6.0,用大孔非离子吸收树脂(Diaion HP-20)柱层析,用水洗脱。在冷却下,用10%盐酸将流出液酸化至Ph=2.3。过滤收集沉淀,干燥,得3.55g头孢克肟,收率34.1%。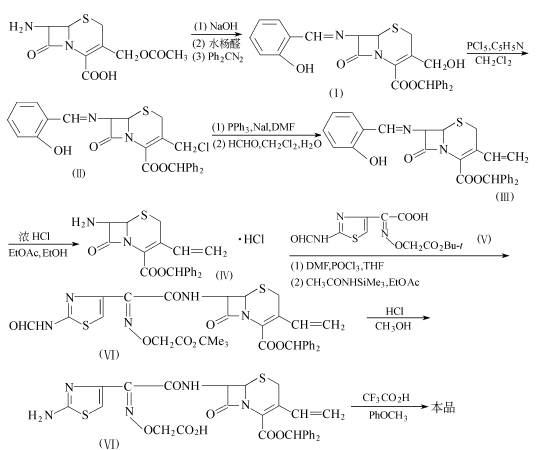
方法2:以脱乙酰头孢菌素C为原料。
脱乙酰头孢菌素C钠盐(118.6g,0.3mo1)溶于1.0L水和0.6L丙酮,在10~15℃下加入苯甲酰氯(42.1g,0.3mo1),在加入过程中,用20%碳酸钠溶液保持反应液的Ph值在6.5~7.5。加毕,在10~15℃下搅拌1h。减压蒸去丙酮,剩下的水溶液用乙酸乙酯洗。往水溶液中加入300ml乙酸乙酯,加入重氮二苯甲烷(135.8g,0.75mo1)在1.0L乙酸乙酯中的溶液。用浓盐酸调至Ph=3.5后,在室温下搅拌1.5h。再用浓盐酸将反应液调至Ph=2.5后,分出有机层,用盐水洗,干燥,减压浓缩。剩余物溶于400ml丙酮,加入4.0L异丙醚。收集沉淀,得224.8g化合物(Ⅷ),收率92.5%。
化台物(Ⅷ)(100g,0.123mo1)溶于600ml二氯甲烷,在-30℃下加入五氯化磷(25.6g,0.123mo1),再加入吡啶(9.8g,0.123mo1)。在-20~10℃下搅拌1h后,将反应液倾入500ml二氯甲烷和300ml水的混合液中。分出二氯甲烷层,用盐水洗,干燥,减压l浓缩。剩余物用异丙醚浸渍,得114.5g粉末状的化合物(Ⅸ),收率98%。
化合物(Ⅸ)(102g,0.123mo1)溶于300ml二甲基甲酰胺,在35℃下加入碘化钠(18.4g,0.123mo1),搅拌30min。加入三苯膦(48.5g,0.185mo1),在35~38℃下搅拌1h。减压浓缩至剩2/3体积后,将其滴加到5.0L异丙醇中。过滤收集沉淀,用异丙醚洗,干燥。减压浓缩,剩余的固体约123.5g溶于1.0L二氯甲烷,在25℃下加入300ml 36%甲醛,搅拌2h,并用20%碳酸钠保持Ph值为9.0。反应结束后,用10%盐酸调至Ph=5.0。分出有机层,用盐水洗,减压蒸发。剩余物用乙酸乙酯浸渍,得63.5g粉状末的化合物(X),收率75.8%。
在5℃下,往五氯化磷(15.5g,74.4mmo1)在200ml二氯甲烷的溶液中,加入吡啶(5.9g,74.4mmo1),搅拌20min。在5℃下加入化合物(X),搅拌2h,冷至-40℃,一次性加入同样冷至-40℃的120ml甲醇。反应液在1h内缓慢升至20℃。反应液减压浓缩,剩余物用300ml乙酸乙酯和50ml水浸渍。过滤收集沉淀,用50ml异丙醇洗2次,再用50ml异丙醚洗。得8.4g结晶状的化合物(Ⅳ),收率79.0%。
化合物(Ⅳ)再和方法1一样反应得到头孢克肟。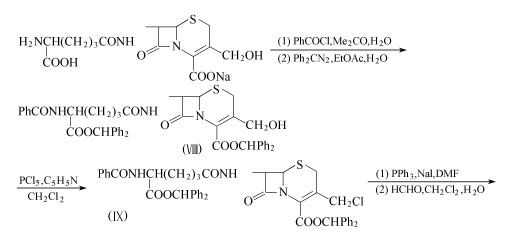
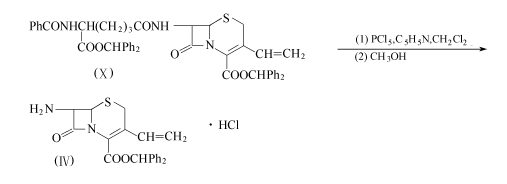 侧链的制备:以乙酰乙酸乙酯为原料,在乙酸中亚硝化在2位引入羟肟,用硫酰氯氯化在4位引入氯,和硫尿环合得到噻唑衍生物,和三苯甲基氯反应以保护2位上的氨基,再和溴乙酸叔丁酯反应,以在肟羟基的氧上引入乙酸基,氢氧化钠水解再氯化使成酰氯,即得到所需的噻唑侧链。
侧链的制备:以乙酰乙酸乙酯为原料,在乙酸中亚硝化在2位引入羟肟,用硫酰氯氯化在4位引入氯,和硫尿环合得到噻唑衍生物,和三苯甲基氯反应以保护2位上的氨基,再和溴乙酸叔丁酯反应,以在肟羟基的氧上引入乙酸基,氢氧化钠水解再氯化使成酰氯,即得到所需的噻唑侧链。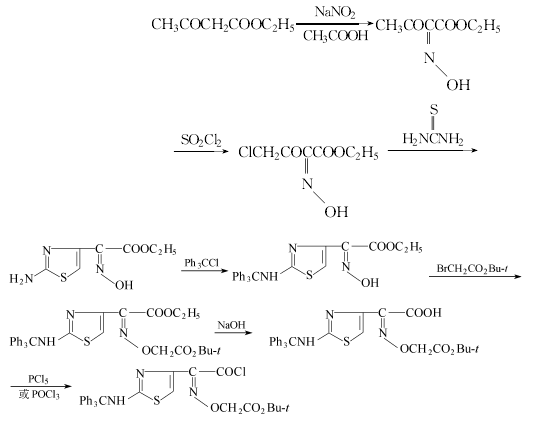
| 危害码 (欧洲) | Xn: Harmful; |
|---|---|
| 风险声明 (欧洲) | R42/43 |
| 安全声明 (欧洲) | S22-S36/37-S45 |
| WGK德国 | 3 |
| 海关编码 | 2941905990 |








