


 搜索
搜索



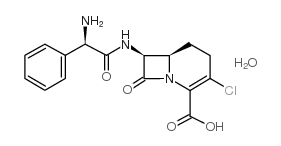
氯碳头孢
-
中文名7-[(氨苯基乙酰基)氨基]-3-氯-8-氧代-1-氮杂二环[4.2.0]辛-2-烯-2-羧酸水合物
-
英文名Loracarbef hydrate
-
中文别名罗拉碳头孢
-
英文别名
1-Azabicyclo4.2.0oct-2-ene-2-carboxylic acid,7-(2R)-aminophenylacetylamino-3-chloro-8-oxo-,monohydrate,(6R,7S) kt3777 Carbacefaclor 7-((aminophenylacetyl)amino)-3-1-azabicyclo(4.2.0)oct-2-ene-2-carboxylicaci -
常用名氯碳头孢
-
C A S号121961-22-6
-
密度N/A
-
沸点662.2ºC at 760 mmHg
-
熔点205-215° (dec) (Matsukuma)
-
化学式C16H18ClN3O5
-
结构式
1、 摩尔折射率:86.23
2、 摩尔体积(cm3/mol):228.8
3、 等张比容(90.2K):228.8
4、 表面张力(dyne/cm):75.7
5、 极化率(10-24cm3):34.18
-
闪点354.3ºC
-
分子量367.78400
-
精确质量367.09300
-
P S A121.96000
-
外观形状
-
储存条件
-
稳定性
-
水溶解性
-
形态
-
H L B值
-
粘度
-
PH值
方法1:528mg化合物(I)溶于15ml无水苯,加入0.2rnl苯硫酚和0.2ml哌啶,在室温下搅拌2h。用10%柠檬酸水溶液和饱和盐水洗,干燥,减压浓缩。油状的剩余物用硅胶柱层析,己烷-乙酸乙酯(4:1)洗脱,得720mg化合物(Ⅱ),收率96.3%,熔点77.5~78℃。
480mg化合物(Ⅱ)溶于50ml蒸馏过的氯仿,在冰浴冷却下,加入240mg间氯过苯甲酸(m-CPBA)。在0℃下搅拌30min后,反应液用饱和碳酸氢钠溶液和饱和盐水洗,干燥,浓缩得500mg化合物(Ⅲ),收率99.9%,熔点95.5~96.5℃。
109mg化合物(Ⅲ)和23.5mg氧化钙悬浮于1ml二氯甲烷,在0℃下加入27μl硫酰氯。并在0℃下搅拌1h。反应液依次用10%柠檬酸溶液、饱和碳酸氢钠和饱和盐水洗,干燥,浓缩。剩余物用硅胶柱层析,己烷-乙酸乙酯(5:1)洗脱,得66.5mg油状的化合物(Ⅳ),收率56.1%。
1.3g化合物(Ⅳ)溶于100ml四氯化碳,回流6h。冷却后,将反应液浓缩。剩余物用硅胶柱层析,正己烷-乙酸乙酯(5:1)洗脱,得596mg化合物(V),收率65.2%,熔点96.0~97.0℃。
350mg化合物(V)和70mg 10%钯-炭催化剂悬浮于100ml乙醇和1.2ml1mol/L盐酸,在室温和常压下,氢化3h,,滤去催化剂,并用小量乙醇洗。滤液和洗液合并后浓缩。剩余的固体溶于水,并用乙醚洗。水层的Ph值用碳酸氢钠调至8后,用乙酸乙酯提取。提取液用饱和盐水洗,干燥,浓缩,得218mg无色粉末状的化合物(Ⅵ),收率68.4%。
102mg化合物(Ⅵ)和1ml三氟乙酸在室温下反应30min。浓缩后,剩余物用乙醚浸渍,得75.5mg黄色粉末状的化合物(Ⅶ),收率75.5%。
约1mm直径的多孔陶瓷柱经γ-氨基丙基三乙氧硅烷处理后,再用谷氨醛处理。粗酶(Kluyvera citrophila KY-7844的提取物)和上述处理过的多孔陶瓷反应,得到固定酶。将9.73g化合物(Ⅶ)溶于580ml磷酸缓冲液(Ph=6.75)中,加入51g苯甘氨酸甲酯。该混合液通过上述制备的含固定酶(185m1)的柱子,柱子的内温保持在20℃,循环3次,约8.5h。可得6.15g氯碳头孢,收率78.2%,熔点205~215℃(分解)。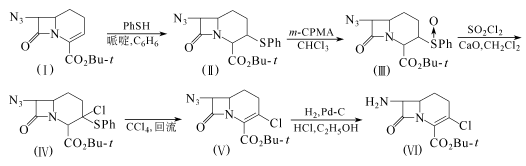
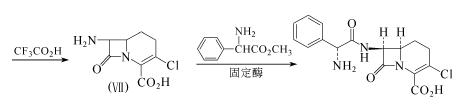 方法2:9.20g化合物(Ⅷ)溶于100ml氯仿,加入2.83ml苯硫酚和0.5ml哌嗪,搅拌3h。浓缩,剩余物用硅胶柱层析,正己烷-乙酸乙酯(2:1)洗脱。得9.26g无色的化合物(Ⅸ)结晶,收率77.5%。
方法2:9.20g化合物(Ⅷ)溶于100ml氯仿,加入2.83ml苯硫酚和0.5ml哌嗪,搅拌3h。浓缩,剩余物用硅胶柱层析,正己烷-乙酸乙酯(2:1)洗脱。得9.26g无色的化合物(Ⅸ)结晶,收率77.5%。
4.78g化合物(Ⅸ)溶于100ml氯仿,在0℃下加入2.37g问氯过氧苯甲酸,搅拌30min后,再加入1 ml10%硫代硫酸钠溶液。分出有机层,用饱和碳酸氢钠和饱和盐水洗,干燥,浓缩。剩余的白色固体溶于50ml二氯甲烷,在冰浴冷却下加入1.45ml硫酰氯,在0℃下搅拌1h。用50ml二氯甲烷稀释后,依次用10%柠檬酸溶液、碳酸氢钠溶液和饱和盐水洗,干燥,浓缩。得到的无色粉末溶于80ml甲苯,回流2h后浓缩。剩余物用硅胶层析,用正己烷-乙酸乙酯(1:1)洗脱,得3.50g无色的化合物(X)的结晶,收率87%,熔点177.6℃。
1.7g化合物(X)和8ml三氟乙酸在0℃下搅拌1h。减压浓缩后,得一棕色油。用乙醚浸渍后得1.1g化合物(Ⅺ),收率71.8%。
10.96g化合物(Ⅺ)溶于150ml水,用1.85倍的水合肼处理后,用0.2mol/L氢氧化钠调至Ph=7.5~7.7。在5℃下搅拌3h后,升温至35℃,用1mol/L盐酸酸化至Ph=0.8,放置4h。过滤除去沉淀,滤液浓缩至93ml,在室温放置过夜。过滤收集析出的沉淀,干燥后得5.54g化合物(Ⅶ)的粗品,纯度83.5%,收率70.6%。用层析提纯后可得无色的化合物(Ⅶ)的结晶。
化合物(Ⅶ)可按方法1得到氯碳头孢。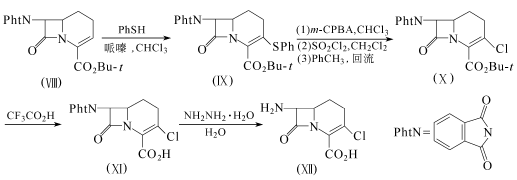
| 危险品运输编码 | NONH for all modes of transport |
|---|








