


 搜索
搜索



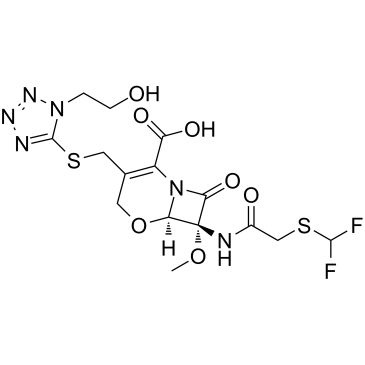
氟氧头孢
-
中文名氟氧头孢
-
英文名Flomoxef
-
中文别名氟莫头孢 | (6R-CIS)-7-[[[(二氟甲基)硫基]乙酰基]氨基]-3-[[[1-(2-羟基乙基)-1H-四氮唑-5-基]硫基]甲基]-7-甲氧基-8-氧代-5-氧杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸 | (-)-(6R,7R)-7-(2-((二氟甲基)硫)乙酰氨基)-3-(((1-(2-羟乙基)-1H-四唑-5-基)硫)甲基)-7-甲氧基-8-氧代-5-氧杂-1-氮杂双环[4.2.0]辛-2-烯-2-甲酸
-
英文别名
FMOX Flomoxefum MFCD00865115 Flomoxefo Flomoxef -
常用名氟氧头孢
-
C A S号99665-00-6
-
密度1.9±0.1 g/cm3
-
沸点232-233 °C(lit.)
-
熔点29-31 °C(lit.)
-
化学式C15H18F2N6O7S2
-
结构式
1、 摩尔折射率:106.90
2、 摩尔体积(cm3/mol):267.4
3、 等张比容(90.2K):789.8
4、 表面张力(dyne/cm):76.0
5、 极化率(10-24cm3):42.38
-
闪点>230 °F
-
分子量496.466
-
精确质量496.064636
-
P S A219.60000
-
外观形状
-
储存条件
-
稳定性
-
水溶解性
-
形态
-
H L B值
-
粘度
-
PH值
往由25.3g钠和700rnl无水乙醇制得的乙醇钠溶液中,加入109.6ml巯基乙酸乙酯,在室温下快速通入氟利昂22进行鼓泡,当温度升至60℃时,调慢通入的速度,并在40℃下继续通入2h。反应液用浓盐酸(9m1)中和,减压蒸出乙醇。往剩余物中加入1000ml乙酸乙酯、50ml lmol/L氢氧化钠和冰水。分出的水层用500ml乙酸乙酯提取。提取液和有机层合并,用水和饱和氯化钠水溶液洗,无水硫酸镁干燥。蒸去溶剂,真空蒸馏得119.8g化合物(I),收率70.5%,沸点46—48.5℃(400Pa)。
381.4g化合物(I)和491ml 25%氢氧化钾水溶液,在冰浴冷却下剧烈搅拌2h,缓慢形成均匀的溶液。反应液和300ml乙酸乙酯及100ml饱和氯化钠水溶液混合。分出有机层,用100ml水洗,水层用300ml乙酸乙酯提取。所有水层合并,和1.6L乙酸乙酯混合,用255ml浓盐酸酸化,再用270g氯化钠使其饱和。分出有机层,用饱和氯化钠水溶液洗,无水硫酸镁干燥。减压浓缩,得309.3g化合物(Ⅱ),收率97.1%。无需提纯,可直接用于以后的反应。 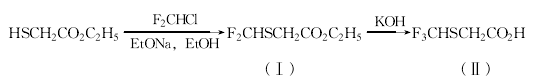 30.54g乙醇胺溶于370ml乙醇和30ml水中,在15℃加入60.6g三乙胺和45g二硫化碳。搅拌lh后,再在15℃加入80g碘甲烷。搅拌30min后,反应液减压浓缩,用350ml水和己烷稀释,放置分层。分出水层,用1.5ml磷酸酸化。用乙酸乙酯提取,提取液浓缩,得81.7g含小量溶剂的化合物(III),无需提纯,直接用于下步反应。
30.54g乙醇胺溶于370ml乙醇和30ml水中,在15℃加入60.6g三乙胺和45g二硫化碳。搅拌lh后,再在15℃加入80g碘甲烷。搅拌30min后,反应液减压浓缩,用350ml水和己烷稀释,放置分层。分出水层,用1.5ml磷酸酸化。用乙酸乙酯提取,提取液浓缩,得81.7g含小量溶剂的化合物(III),无需提纯,直接用于下步反应。
将上述得到的化合物(III)溶于300ml二氯甲烷,加入54g二氢吡喃和1.0g对甲苯磺酸单水合物,搅拌lh。反应液用碳酸氢钠水溶液洗,浓缩得129g粗品化合物(IV)。
11.8g化合物(IV)溶于40ml乙醇,加入含3.40g叠氮钠的20ml水溶液,回流2.5h。反应液减压浓缩,剩余物溶入水,用乙酸乙酯提取后,用磷酸酸化,再用乙酸乙酯提取。提取液水洗,干燥,浓缩得10.4g固体。该固体为化合物(V)的吡咯醚。将该固体溶于丙酮水溶液,用浓盐酸酸化至Ph=2,在室温下放置2h。浓缩,用乙酸乙酯提取。提取液干燥,浓缩。剩余物用乙酸乙酯和己烷的混合溶剂重结晶,得6.2g化合物(V),收率85%,熔点135~137℃。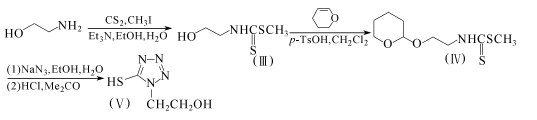
977mg化合物(V)溶于5ml二甲基甲酰胺,在-20*℃下加入1.3ml 5.2mol/L甲醇钠的甲醇溶液。在-20℃下,把该溶液加到2.80g化合物(Ⅵ)(其制备参见Yoshioka M,et a1.Tetrahedron Lett,1980,21:351 354)在19ml二甲基甲酰胺中的溶液。加毕,搅拌2h。反应液倾入冰水,用乙酸乙酯提取。提取液水洗,无水硫酸钠干燥,浓缩至于。剩余物用硅胶层析,得3.7g含小量溶剂的化合物(Ⅶ),收率几乎定量。
3.38g化合物(Ⅶ)和3.6ml吡啶溶于3ml二氯甲烷,在一20℃下加入3.4g对甲基氯甲酸苄酯在2ml二氯甲烷中的溶液,搅拌5h。反应液倾入冷水,常规处理后用硅胶层析,得3.2g化合物(Ⅷ),收率82.6%(以化合物(V)计)。
1.0g化合物(V1)溶于10ml二氯甲烷,冷至-55℃,依次和192μl次氯酸叔丁酯及0.92ml 2mol/L甲醇锂的甲醇溶液反应。形成的溶液搅拌15min后,加入0.8ml乙酸以终止反应。反应液倾人冰水,用乙酸乙酯提取。提取液常规处理后用硅胶层析,得770mg化合物(Ⅸ),收率74%。
500mg化合物(Ⅸ)溶于10ml二氯甲烷,在冰浴冷却下,依次加入112μl吡啶和263mg五氯化磷。加毕,反应液升至室温,搅拌2.5h。冷至-20℃,加入5ml甲醇。在2~3℃下再搅拌3h。反应液倾入冷的碳酸氢钠水溶液,用乙酸乙酯提取。提取液常规处理,得326mg化合物(X)的结晶,收率75%,熔点135~138.5℃。该化合物对空气稍敏感。
1.0g化合物(X)悬浮于二氯甲烷,在冰浴冷却下,加入0.52g吡啶和0.17g化合物(Ⅱ)。冷至-15℃,缓慢加入0.25g三氯氧磷。所成溶液在-17~-15℃下搅拌20min后,倾人冰水,用二氯甲烷提取。提取液用水洗3次,无水硫酸钠干燥,减压浓缩。剩余物用乙酸乙酯浸渍后得935mg化合物(Ⅺ)的结晶,收率83%,熔点80~82℃。
405rng化合物(Ⅺ)、2.5ml二氯甲烷和0.5ml硝基甲烷混合,在-30℃下加入0.11 ml苯甲醚和0.17ml四氯化锡在2ml二氯甲烷中的溶液。在搅拌的3.5h中,缓慢升温至-IO℃,倾人1mol/L盐酸、乙酸乙酯和甲乙酮的混合液中。分出的有机层和碳酸氢钠水溶液混合,水层用浓盐酸酸化,用乙酸乙酯和甲乙酮的混合液来提取。提取液用盐水洗,无水硫酸镁干燥,浓缩。剩余物用丙酮和二氯甲烷结晶,得260mg结晶状的氟氧头孢,收率100%,熔点82.5~87.5℃。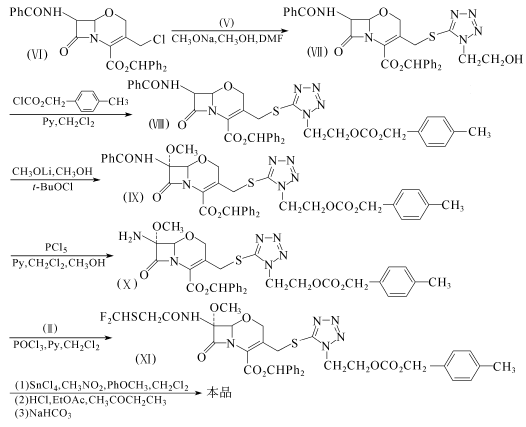
| 危害码 (欧洲) | Xi |
|---|---|
| 风险声明 (欧洲) | R37/38:Irritating to respiratory system and skin . R41:Risk of serious damage to eyes. R36/37/38:Irritating to eyes, respiratory system and skin . |
| 安全声明 (欧洲) | S26-S36-S37/39 |
| WGK德国 | 2 |
| RTECS号 | GU8370000 |








